ACTE III : Du quinquina à la quinine et à ses analogues ou du naturel au synthétique

Le principe actif du remède, la quinine, a été extraite des écorces du quinquina jaune en juin 1820 par deux pharmaciens-chimistes français, Pelletier et Caventou. Cette découverte, très importante à l’époque, est toujours d’actualité en 2020, où nous célèbrerons son bicentenaire. La quinine reste un médicament majeur pour le traitement du paludisme, qui est toujours, avec 219 millions de personnes malades et 435 000 décès en 2017, la parasitose la plus importante. De plus elle concerne majoritairement les enfants de moins de cinq ans.
Depuis 1820 les chimistes ont déterminé la structure moléculaire de la quinine et ont synthétisé cette molécule naturelle relativement complexe et donc coûteuse à produire. Aussi ils se sont efforcés de préparer par la synthèse des analogues moins coûteux, dont la chloroquine objet actuellement de nombreux questionnements.
ACTE III : Du quinquina à la quinine et à ses analogues ou du naturel au synthétique
En 1820 deux jeunes chimistes français, Pierre Joseph Pelletier (1788-1842) et Joseph Bienaimé Caventou (1795-1877), élèves de Nicolas Vauquelin isolent deux alcaloïdes, la quinine et la cinchonine, à partir de l’écorce de quinquina. Leurs recherches permirent à François Magendie, professeur de médecine au Collège de France d’étudier les doses auxquelles ces principes actifs étaient efficaces. Cette découverte marque le passage de l’utilisation de plantes médicinales à celle de leur principe actif, facile à prendre, dosé et caractérisé.
La première tentative de synthèse de la quinine en 1856 est due à William Henry Perkin, un chimiste anglais de 18 ans, alors qu’on n’en connaissait pas encore la structure moléculaire. Il réalise sa synthèse à partir de l’aniline et il n’obtient pas la quinine, mais un autre produit intéressant, la mauvéine. C’est un bon exemple de « découverte faite par hasard dans un esprit préparé » (1). La mauvéine est la première teinture industrielle pour le textile qui résiste au lavage et cette synthèse a révolutionné l’industrie des colorants.
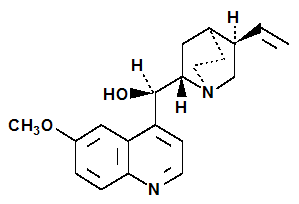
La formule brute de la quinine, C20H24N2O2, a été déterminée par Adolph Strecker en 1854 et la structure moléculaire (2) est due à Zdenko Skraup et Wilhelm Königs à la fin du XIXe siècle.
En 1918, Paul Rabe et Karl Kindler affirment avoir réalisé la synthèse de la quinine à partir de la quinotoxine elle-même isolée de la plante, mais sans donner aucun détail expérimental sur cette hémisynthèse. En 1944, Robert Woodward et William von Eggers Doering annoncent avoir réussi la synthèse totale de la quinine, mais ne décrivent dans leur publication que les 17 étapes conduisant à la quinotoxine et s’en remettent aux travaux de Paul Rabe pour sa transformation en quinine. Cependant la reproduction de cette opération a posé de nombreux problèmes et la synthèse n’a pu être réellement effectuée qu’en 1967 et 1973 par Uskokovic.
La synthèse totale indiscutable et stéréo-sélective a été réalisée par Gilbert Stork en 2001, ce qui généra une controverse avec les auteurs de la synthèse de 1944. Si la synthèse totale de la quinine est d’un grand intérêt scientifique, elle présente peu d’intérêt pratique à cause de son coût, aussi de nombreux essais d’obtention par la synthèse d’analogues simplifiés ont été tentés par les chimistes.
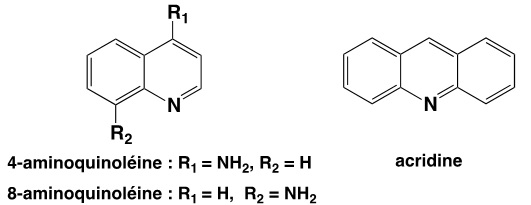
Ainsi, en 1925, la firme germanique Bayer réalise la synthèse de la Plasmoquine (ou Pamaquine) un dérivé de la 8-aminoquinoléine (3) capable d’empêcher les rechutes du paludisme dues à Plasmodium vivax. En 1932, la firme développe un dérivé de l’acridine (3), la Quinacrine (ou Mépacrine) active sur Plasmodium falciparum.
À l’instigation du chimiste Hans Andersag, la firme Bayer développe, parmi 12000 composants synthétiques différents, un composé connu sous le nom de Résoquine, possédant un noyau 4-aminoquinoléine (3) et un de ses dérivés nommé Sontochine (ou 3-méthylrésoquine). Mais bien que jugés prometteurs, ils furent évalués trop toxiques.
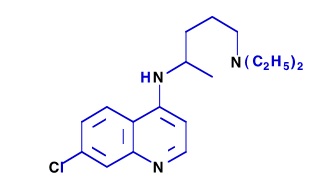
Pendant la Seconde Guerre mondiale, la production de quinine fut interrompue suite à la prise de Java par les Japonais et Plasmoquine et Mépacrine furent alors largement utilisées. Les Américains et Britanniques eurent connaissance de la Résoquine, la synthétisèrent, mais elle fut à nouveau évaluée toxique. Les Français de Vichy travaillaient à Tunis sur la Sontochine. Leurs résultats pharmacologiques récupérés par les Alliés redonnèrent de l’intérêt pour la Résoquine en 1946 qui fut rebaptisée Chloroquine (ou Nivaquine) (4) et qui devint le composé antipaludique le plus efficace et le plus utilisé à travers le monde, jusqu’aux années 1960, où commencèrent les problèmes de résistance du Plasmodium parasite à ce produit.
Mais l’histoire de cette famille de substances naturelles et synthétiques suscitera encore beaucoup d’intérêt en 2020 pour diverses raisons.
Pour en savoir plus - Actes précédents
- ACTE I : Le quinquina, remède du Nouveau Monde pour une maladie de l’Ancien : légendes et réalités d’une découverte
- ACTE II : Le quinquina, polémiques religieuses et querelles médicales
Notes
(1) Citation de Louis Pasteur
(2) Quinine
(3) 8-aminoquinoléine, 4-aminoquinoléine et acridine
(4) Formule de la chloroquine de nom commercial Nivaquine
Pour en savoir plus sur cet acte
- Pelletier et Caventou. Recherches chimiques sur les quinquinas, Ann. Chim. Phys. (1820) 15, p. 289-318
- Robert Woodward et William von Eggers Doering. J. Am. Chem. Soc (1944) 66, p. 849 et J. Am. Chem. Soc (1945) 67, p. 860
- M.R. Uskokovic, J. Gutzwiller, T. Henderson.Total synthesis of quinine and quinidine, J. Am. Chem. Soc, (1970), 92, p. 203-204
- Gilbert Stork, Deqiang Niu, A. Fujimoto, Emil R. Koft, James M. Balkovec, James R. Tata, Gregory R. Dake. The First Stereoselective Total Synthesis of Quinine, J. Am. Chem. Soc. (2001) 123, p. 3239-3242.